CASE REPORT | https://doi.org/10.5005/jp-journals-10006-1791 |
Moschcowitz Syndrome or Thrombotic Thrombocytopenic Purpura and Antiphospholipid Antibody Syndrome as a Rare Cause of Thrombocytopenia in Pregnancy Mimicking Hemolysis, Elevated Liver Enzymes, and Low Platelets Syndrome in a Patient with Bad Obstetric History: A Diagnostic Dilemma
1–3Department of Obstetrics and Gynaecology, Jawaharlal Nehru Medical College, Datta Meghe Institute of Medical Sciences (Deemed University), Sawangi, Wardha, Maharashtra, India
4Department of Medicine, Jawaharlal Nehru Medical College, Datta Meghe Institute of Medical Sciences (Deemed University), Sawangi, Wardha, Maharashtra, India
Corresponding Author: Neema Acharya, Department of Obstetrics and Gynaecology, Jawaharlal Nehru Medical College, Datta Meghe Institute of Medical Sciences (Deemed University), Sawangi, Wardha, Maharashtra, India, Phone: +91 9326692511, e-mail: neemasacharya@gmail.com
How to cite this article Mohammad S, Bhute A, Acharya N, et al. Moschcowitz Syndrome or Thrombotic Thrombocytopenic Purpura and Antiphospholipid Antibody Syndrome as a Rare Cause of Thrombocytopenia in Pregnancy Mimicking Hemolysis, Elevated Liver Enzymes, and Low Platelets Syndrome in a Patient with Bad Obstetric History: A Diagnostic Dilemma. J South Asian Feder Obst Gynae 2020;12(4):250–253.
Source of support: Nil
Conflict of interest: None
ABSTRACT
Thrombotic microangiopathy (TMA) is a group of disorders characterized by microangiopathic hemolytic anemia, moderate to severe thrombocytopenia, and end-organ damage. In pregnancy, TMA is most commonly due to preeclampsia and hemolysis, elevated liver enzymes, low platelets (HELLP) syndrome. Very rarely it occurs due to Moschowitz syndrome also known as thrombotic thrombocytopenic purpura (TTP), antiphospholipid antibody (APLA) syndrome, or atypical hemolytic uremic syndrome (aHUS). All of these share common clinical features; yet their etiologies are different. Although both HELLP and TTP share common features of endothelial injury and microvascular thrombi, yet, they have different courses and line of management. In term pregnancy, differentiating between these two pathologies can be extremely difficult due to the extreme overlap in clinical and laboratory manifestations, and this becomes possible only with the use of specific markers as ADAMTS-13, whenever available. Unfortunately, delay in timely diagnosis and treatment can be life-threatening. We describe case of 25-year-old pregnant woman who came with HELLP syndrome which did not resolve postpartum only to improve after plasmapheresis.
Keywords: ADAMTS-13, Hemolysis, elevated liver enzymes, low platelets, Plasmapheresis, Preeclampsia, Thrombotic thrombocytopenic purpura..
INTRODUCTION
TTP was first described in 1924 by Eli Moschcowitz and characterized by a pentad of findings that include microangiopathic hemolytic anemia, thrombocytopenia, renal failure, neurologic findings, and fever.
Both hemolysis, elevated liver enzymes, low platelets (HELLP) and thrombotic thrombocytopenic purpura (TTP) share common features of thrombotic microangiopathies, but their pathogenesis is entirely different. So, to make a diagnosis early especially in term pregnancy is important to avoid serious fetomaternal complications. Although ADAMTS-13 can discern between both conditions, the results are not readily available, and differential diagnosis needs to be derived relying on clinical judgement. Significance of exact antenatal diagnosis is derived from the fact that two divergent management plans are needed. Optimal treatment plan for TTP is ultimately plasmapheresis and should be instituted as soon as diagnosis is established, while termination of pregnancy does not ameliorate the condition. On the other hand, delivery remains the definitive treatment in HELLP syndrome, whereas plasmapheresis is not a part of usual antenatal management plan. In HELLP/PE, resolution is anticipated within 2–3 weeks, whereas in TTP, the condition will continue to deteriorate unless plasmapheresis is started. Here, once more, the clinical condition mandates plasmapheresis irrespective of the identity of the offending disorder, pending the results of ADAMTS-13.1 Hereby, we describe the clinical course and evolution of laboratory values encountered in a pregnancy, where Moschcowitz syndrome (TTP) was mimicking as HELLP syndrome. In addition, we also present a practical plan for the differential diagnosis and management of HELLP syndrome and TTP.
CASE DESCRIPTION
A 25-year-old third gravida para two, no live issues, with 34 weeks POG was admitted to hospital with complaints of headache since 2 days and three episodes of convulsions a day before. Cardiotocography on admission was non-reassuring with a saltatory pattern. Her blood pressure was 180/110 mm Hg at the time of admission. The patient was drowsy, icteric, and pale with minimal bilateral pedal edema. Her first pregnancy resulted in spontaneous abortion at 16 weeks, second pregnancy continued till 27 weeks, and was terminated in view of severe preeclampsia. APLA tested positive during this pregnancy. Third pregnancy was present pregnancy, and she had history of hypertension since first trimester. After administering labetalol and stabilizing BP, she was taken was for emergency cesarean section indicated in view of HELLP syndrome with severe oligohydramnios with absent diastolic flow in umbilical artery on fetal color Doppler. Intraoperative liquor seemed very less, and a female baby, small for gestation, was extracted by vertex presentation. Baby was handed over to neonatologist in view of delayed cry. Placenta was anterior, and there were signs of placental separation. The placenta and umbilical cord looked abnormal which was confirmed by the presence of multiple placental and umbilical cord thrombi on pathology (Fig. 1). Placenta and membranes expelled, and abdomen was closed in layers. Uterus was relaxing in-between so Pitocin 10/20 units started ivf, massage given, inj carboprost given intramyometrial. Uterus retracted well. Skin closed by mattress suture. Her vitals showed blood pressure of 200/100 mm Hg, heart rate of 92 beats per minute, respiratory rate of 32 breaths per minute, and temperature of 98.3°F. On physical examination, she was in no acute distress, heart examination showed tachycardia with normal sinus rhythm, normal heart sounds, and lungs were clear on auscultation bilaterally. There were no signs of focal deficit on neurological examination.
Her labs on admission showed a white cell count 14,500/mm3, hemoglobin 7.7 g/dL, and platelets 27,000/mm3 compared to normal counts. There were few schistocytes on peripheral blood smear. Sickling was negative. Markers of hemolysis were positive (reticulocytes 6.2%, lactate dehydrogenase (LDH) 1190 U/L with upward trend thereafter, haptoglobin %3C;8 mg/dL). Direct Coomb’s test was negative. Prothrombin time (PT), activated partial thromboplastin time (aPTT), and fibrinogen were normal. D-dimers were elevated at %3E;2,000 ng/mL. The patient was HIV negative.
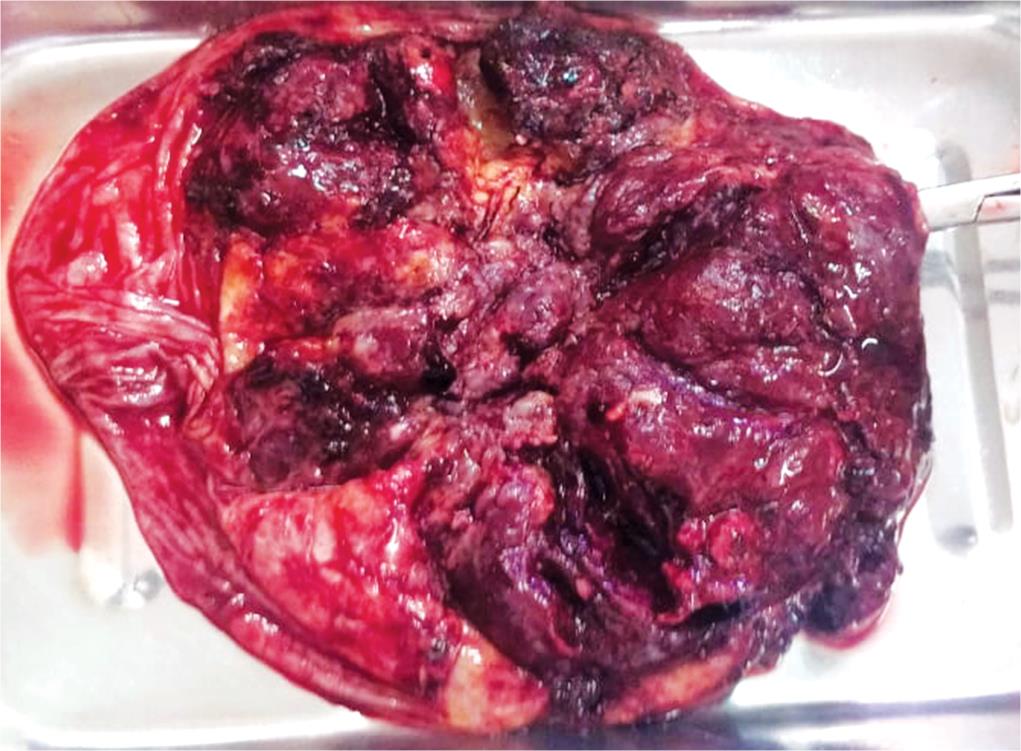
Fig. 1: Small infarcts of dark red and granular appearance
Postoperative hemoglobin was 5.5 g/dL, white cell count 12,300/mm3, and platelets 14,000/mm3.
Serial liver enzymes and liver function tests worsened. Her urinalysis had significant proteinuria. Additional testing for antiphospholipid
Antibodies were positive, and platelet factor 4 antibodies (HPF4) were negative. Table 1 shows trends of her platelets, AST, and LDH. On POD #4, she was shifted to intensive care unit where she was managed with anticonvulsive and antihypertensive medications and other supportive treatment like BIPAP. SDP and PRC were transfused accordingly. Plasmapheresis 7 sessions were done for consecutive days. The patient then stabilized after plasmapheresis, with reducing LDH and improving platelet count and improving renal functions.
The patient was discharged on postpartum day #18 It is to be noted that her LDH to AST ratio on postpartum day #4 was greater than 22 (51.1).
Differential Diagnosis
- TTP/atypical hmeolytic uremic syndrome (aHUS) or Moschowitz syndrome
- Hemolysis, elevated liver enzymes, low platelets (HELLP) syndrome
- Severe preeclampsia
- Disseminated intravascular coagulation (DIC)
- Antiphospholipid antibody syndrome (APLA).
DISCUSSION
In our patient, there was an evidence of hemolysis with positive markers like high reticulocyte count, low haptoglobin, and high LDH in addition to Negative Coomb’s test, severe thrombocytopenia, and placental infarcts that raised a suspicion for a thrombotic microangiopathy. Several microangiopathies can exist in pregnancy, such as preeclampsia/HELLP, complement mediated thrombotic microangiopathy (C-TMA), a-HUS, Moschowitz syndrome also known as thrombotic thrombocytopenic purpura (TTP), antiphospholipid antibody syndrome (APLA), etc. The two most common microangiopathies that are commonly seen in pregnancy are preeclampsia and HELLP. TTP and APLA are rare occurrence. HELLP is a syndrome of MAHA which is characterized by hemolysis, elevated liver enzymes, and low platelet count. Women who do not meet all the criteria are considered to have partial HELLP.2
Initially, our patient was thought to have partial HELLP based on presence of hemolysis, thrombocytopenia, elevated AST, and LDH to AST ratio >22; but since HELLP is related to pregnancy, there should have been rapid and complete resolution after delivery.
Moreover, LDH to AST ratio of >22 is more supportive for diagnosis of TTP over HELLP.3
LDH of patient started trending up, LDH to AST ratio on postpartum day #4 was 51.1, and the clinical condition improved only after plasmapheresis and supports the diagnosis of TTP.
Classical features of TTP consist of the pentad: thrombocytopenia, microangiopathic hemolytic anemia with schistocytes in the blood smear, neurologic abnormalities, fever, and renal failure. First three symptoms occur frequently together (50–75%), but all five symptoms rarely occur together; therefore pentad is considered to be outdated.
Our patient had four of five abnormalities of the pentad of TTP manifested by MAHA, thrombocytopenia, neurological abnormalities (new onset headache and convulsions), and renal failure.
| Hemoglobin (g/dL) | PLTs (mm3) | Reticulocytes (%) | LDH (mg/dL) | Haptoglobin (mg/dL) | AST (U/L) | ALT (U/L) | BUN (mg/dL) | Creatinine (md/dL) | |
|---|---|---|---|---|---|---|---|---|---|
| Postpartum day #0 | 5.5 | 14,000 | 6.2 | 1,190 | 8 | 640 | 355 | 14 | 2.1 |
| Postpartum day #2 | 4.8 | 19,000 | 11.7 | 2,911 | 16 | 168 | 305 | 14 | 3.79 |
| Postpartum day #4 | 8.0 | 25,570 | 12.8 | 1,523 | <6 | 29.8 | 91.7 | 12 | 4.12 |
| Postpartum day #12 | 7.0 | 92,850 | 6.0 | 194 | 18 | 19.8 | 81 | 10 | 0.83 |
The hallmark of TTP is deficiency of an enzyme known as ADAMTS13. It is responsible for the cleavage of the unusually large von Willebrand factor (vWF) multimers.4
Decreased activity of ADAMTS13 leads to persistence of those multimers in the blood, which in turn leads to platelet aggregation and formation of platelet thrombi and hence thrombocytopenia from platelet consumption. Hemolytic anemia and schistocytes result from shearing of RBCs, as they pass through the microthrombi. Microthrombotic occlusion of the microvasculature of different organs (e.g., kidney and brain) results in the myriad of symptoms reported in this condition (confusion, headache, blurred vision, and kidney damage).5
In classic cases, severe deficiency of ADAMTS13 (<5% of normal) is specific for TTP as shown by several cohort studies, where about 33–100% had severe deficiency. This leaves a minority of patients who can still have TTP without being severely deficient in ADAMTS13. However, quick and complete sustained response to plasma exchange confirms the diagnosis of TTP.6
The association between pregnancy and TTP has been reported in several case series in the literature. It has been suggested that the physiological changes that take place during pregnancy can precipitate TTP in the susceptible individual. Those changes include a hypercoagulable state with 1.5-fold to 3-fold increase in von Willebrand factor among others with a parallel progressive decrease in ADAMTS13. In other words, the higher the vWF concentration, the more ADAMTS13 will be consumed in breaking down its multimers. In a susceptible individual with either congenital or autoimmune-mediated deficiency of ADAMTS13, this fine balance is tipped, resulting in more vWF multimers beyond the cleavage capacity of ADAMT13.7,8
The two main types of TTP are congenital and acquired. The congenital type can present at any age. However, most patients have delayed onset in the childbearing age when subjected to a triggering event. Common precipitating factors are infection (e.g., influenza), pregnancy, surgery, and pancreatitis.9
Of note, a first episode of pregnancy-related TTP could be congenital and is confirmed by the detection of mutation in the ADAMTS13 gene. The acquired type is autoimmune mediated. The diagnosis is made by identifying ADAMTS13 IgG antibodies in serum.10 However, no further testing was pursued in our patient to identify her specific subtype.
Plasma exchange should be promptly initiated once diagnosis of TTP is suspected. In certain conditions, where there is overlap of clinical manifestations between different microangiopathies, a decision could be made to proceed with plasma exchange based on high index of suspicion.4
Plasma exchange serves two purposes, first removal of the plasma along with all the preformed microthrombi and autoantibodies and second is repletion of the deficient ADAMTS13 activity by administration of plasma rich in ADAMTS13. Hence, it is superior over plasma infusion which can serve only the latter purpose and is accompanied by a risk of volume overload.11
Rituximab, the anti-CD20 antibody, is safe and effective treatment for newly diagnosed TTP along with plasma exchange. It has been shown to decrease the number of plasma exchanges required to achieve remission, to decrease the length of inpatient stay, and to reduce the risk of relapse by over 80%. In addition to its use in acute TTP, it may be used preemptively to prevent relapse of TTP.12
Platelet transfusion is another controversial topic in TTP. Current guidelines advise against routine platelet transfusion in TTP for fear of fueling the fire, except in the setting of severe bleeding or the need for invasive procedures. The most recent data from a retrospective study showed that platelet transfusions were associated with higher odds of arterial thrombosis and mortality among patients with TTP.12
It also showed that pregnancy outcomes have been favorable once early detection and preemptive therapy were begun. The approach that they adopted for cTTP in subsequent pregnancies consisted of regular plasma infusions from 8 to 10 weeks’ gestation every 2 weeks, increased to weekly from 20 weeks gestation until delivery. For acquired TTP, ADAMTS13 level at the onset of subsequent pregnancies is a good predictor of the risk of relapse.9 If ADAMTS13 activity falls to <10% at any point, elective plasma exchange should be started and continued through term 23. When low ADAMTS13 preceded pregnancy, elective rituximab 12 weeks before conception was associated with successful pregnancy outcomes per the UK TTP registry.13
ADAMTS13 activity should be measured in all patients in who the diagnosis of TTP is suspected. However, because the results of ADAMTS13 activity measurements may not be available for several days, and also because of potential variability among assays, the initial diagnosis of TTP and the decision to begin plasma exchange treatment is based on the clinical features. Using these diagnostic criteria, the presence of PE/HELLP syndrome could exclude the diagnosis of TTP by providing an alternative etiology for MAHA and thrombocytopenia. However, the diagnosis of TTP must be considered in addition to PE/HELLP syndrome. The frequency of TTP is increased in women during pregnancy and postpartum because pregnancy can precipitate an acute episode.14
CONCLUSION
TTP can be life-threatening in term pregnancy if not diagnosed and managed timely. Therefore, diagnosis of TTP is of utmost importance. Due to major overlapping of features of TTP with APLA and other thrombotic microangiopathies, diagnosis becomes a diagnostic dilemma. Very low ADAMTS-13 titer confirms the diagnosis of TTP. However, as it is not easily available for our patient, improvement after plasmapheresis confirmed the diagnosis. Also, it is important to note that there exists a continuum between these thrombotic microangiopathies such as APLA and TTP as in the case of our patient.
REFERENCES
1. ADAMTS-13 in the Diagnosis and Management of Thrombotic Microangiopathies [WWW Document], n.d. URL https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4222415/(accessed 5.30.20).
2. Audibert F, Friedman SA, Frangieh AY, et al. Clinical utility of strict diagnostic criteria for the HELLP (hemolysis, elevated liver enzymes, and low platelets) syndrome. Am J Obstet Gynecol 1996;175(2):460–464. DOI: 10.1016/S0002-9378(96)70162-X.
3. Keiser SD, Boyd KW, Rehberg JF, et al. A high LDH to AST ratio helps to differentiate pregnancy-associated thrombotic thrombocytopenic purpura (TTP) from HELLP syndrome. J. Matern-fetal Neonatal Med 2012;25:1059–1063. DOI: 10.3109/14767058.2011.619603.
4. Elayoubi J, Donthireddy K, Nemakayala DR. Microangiopathies in pregnancy. BMJ Case Rep 2018;2018:bcr2017221648. DOI: 10.1136/bcr-2017-221648.
5. Levy GG, Nichols WC, Lian EC, et al. Mutations in a member of the ADAMTS gene family cause thrombotic thrombocytopenic purpura. Nature 2001;413(6855):488–494. DOI: 10.1038/35097008.
6. Sarig G. ADAMTS-13 in the diagnosis and management of thrombotic microangiopathies. Rambam Maimonides Med J 2014;5(4):e0026. DOI: 10.5041/RMMJ.10160.
7. Ducloy-Bouthors AS, Caron C, Subtil D, et al. Thrombotic thrombocytopenic purpura: medical and biological monitoring of six pregnancies. Eur J Obstet Gynecol Reprod Biol 2003;111(2):146–152. DOI: 10.1016/S0301-2115(03)00199-4.
8. Martin JN, Bailey AP, Rehberg JF, et al. Thrombotic thrombocytopenic purpura in 166 pregnancies: 1955–2006. Am J Obstet Gynecol 2008;199(2):98–104. DOI: 10.1016/j.ajog.2008.03.011.
9. Fujimura Y, Matsumoto M, Isonishi A, et al. Natural history of Upshaw-Schulman syndrome based on ADAMTS13 gene analysis in japan. J Thromb Haemost 2011 (9 Suppl 1):283–301. DOI: 10.1111/j.1538-7836.2011.04341.x.
10. Caeiro Alves F, Aguiar R, Pessegueiro P, et al. Thrombotic microangiopathy associated with mycoplasma pneumoniae infection. BMJ Case Rep 2018;2018:bcr2017222582. DOI: 10.1136/bcr-2017-222582.
11. George JN. How I treat patients with thrombotic thrombocytopenic purpura: 2010. Blood 2010;116(20):4060–4069. DOI: 10.1182/blood-2010-07-271445.
12. Goel R, Ness PM, Takemoto CM, et al. Platelet transfusions in platelet consumptive disorders are associated with arterial thrombosis and in-hospital mortality. Blood 2015;125(9):1470–1476. DOI: 10.1182/blood-2014-10-605493.
13. Scully M, Thomas M, Underwood M, et al. Thrombotic thrombocytopenic purpura and pregnancy: presentation, management, and subsequent pregnancy outcomes. Blood 2014;124(2):211–219. DOI: 10.1182/blood-2014-02-553131.
14. George JN, Nester CM, McIntosh JJ. Syndromes of thrombotic microangiopathy associated with pregnancy. Hematology 2015;2015:644–648. DOI: 10.1182/asheducation-2015.1.644.
________________________
© The Author(s). 2020 Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by-nc/4.0/), which permits unrestricted use, distribution, and non-commercial reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.